Εισαγωγή
Το αιμοφαγοκυτταρικό σύνδρομο, αλλιώς γνωστό και ως αιμοφαγοκυτταρική λεμφοϊστιοκυττάρωση (Hemophagocytic Lymphohistiocytosis- HLH), αποτελεί μια απειλητική για τη ζωή κατάσταση, η οποία περιγράφηκε για πρώτη φορά το 1939 από τους Παιδιάτρους Ronal Scott και Robb-Smith. Πρόκειται για ένα κλινικό σύνδρομο υπερ φλεγμονώδους αντίδρασης, το οποίο συνοδεύεται από υπερέκκριση (καταρράκτη) κυτταροκινών ως αποτέλεσμα του ισχυρά διεγερμένου, αλλά αναποτελεσματικού ανοσοποιητικού συστήματος.

Κατατάσσεται στην ευρύτερη ομάδα των ιστιοκυτταρώσεων (βλ. πίνακα), στις οποίες κοινός παρονομαστής είναι ο πολλαπλασιασμός και η συσσώρευση δενδριτικών κυττάρων και μακροφάγων στα σημεία των βλαβών.
| Διαταραχές Δενδριτικών κυττάρων | Ιστιοκυττάρωση Langerhans (LCH)Νεανικό Ξανθοκοκκίωμα (JXG)Νόσος Erdheim-Chester (ECD) |
| Διαταραχές σχετιζόμενες με Mακροφάγα | Αιμοφαγοκυτταρική λεμφοϊστιοκυττάρωση (HLH)Νόσος Rosai-Dorfman (RDD) |
| Κακοήθεις ιστιοκυτταρικές διαταραχές | ΛευχαιμίεςΚακοήθεις όγκοι |
Ορισμός
Η αιμοφαγοκυτταρική λεμφοϊστιοκυττάρωση (HLH), ορίζεται ως σύνδρομο παθολογικής υπεράνοσης αντίδρασης με κλινικά σημεία και συμπτώματα ως επί βαρείας φλεγμονώδους αντίδρασης.
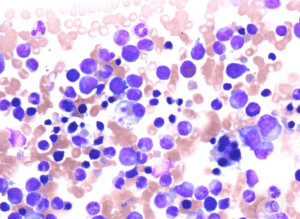
Επιπλέον, θα πρέπει να διαφοροποιείται από την αιμοφαγοκυττάρωση, η οποία αποτελεί ιστολογικό εύρημα και χαρακτηρίζει τη φαγοκυττάρωση ερυθρών αιμοσφαιρίων, λευκών αιμοσφαιρίων, αιμοπεταλίων, καθώς και των πρόδρομων μορφών τους στο μυελό των οστών ή/και σε άλλους ιστούς από μακροφάγα κύτταρα, ιστιοκύτταρα ή άλλα κύτταρα του δικτυοενδοθηλιακού συστήματος.



https://www.pathologyoutlines.com/topic/bonemarrowhemophagocyticlymphohistiocytosis.html
Γιατί λέγεται αιμοφαγοκυτταρικό; Γιατί όπως είπαμε, ιστολογικά, χαρακτηρίζεται από φαγοκυττάρωση από τα μακροφάγα ερυθρών (αιμο-) αιμοσφαιρίων.
Επιδημιολογία
Ο ακριβής υπολογισμός της επίπτωσης και της κατανομής της HLH είναι δύσκολο να επιτευχθεί, αφενός λόγω των μη ειδικών κριτηρίων που απαιτούνται για τη διάγνωση και αφετέρου λόγω της συνοσηρότητας που εμφανίζουν πολλοί ασθενείς τη στιγμή της διάγνωσης. Η πιο ολοκληρωμένη παρουσίαση επιδημιολογικών δεδομένων, αφορά σε παιδιατρικά περιστατικά πρωτοπαθούς HLH κατά τα έτη 1987-2006 στη Σουηδία, με υπολογιζόμενη ετήσια επίπτωση ~ 1.5 περίπτωσεις ανά 1 εκατομμύριο πληθυσμού και πιο ειδικά, ~ 1 περίπτωση ανά 3.000 εισαγωγές σε παιδιατρικό τριτοβάθμιο νοσοκομείο.
Στους ενήλικες με δευτεροπαθή HLH, ακριβείς υπολογισμοί είναι ακόμη δυσκολότερο να επιτευχθούν. Αυτό συμβαίνει διότι η κλινική εικόνα του συνδρόμου είναι δύσκολο να διακριθεί από την αντίστοιχη του πρωτοπαθούς αιτίου (π.χ. σήψη, διάχυτη μεταστατική νόσος, έξαρση αυτοάνοσου νοσήματος) και επιπλέον, ο εργαστηριακός έλεγχος που απαιτείται για την πλήρωση των κριτηρίων περιστασιακά παραλείπεται. Μερικές μελέτες, αναφέρουν ότι η ετήσια επίπτωση είναι ~ 1 περίπτωση ανά 2.000 εισαγωγές ασθενών σε μονάδες εντατικής θεραπείας. Ο βαθμός κλινικής υποψίας και ο τρόπος λειτουργίας κάθε κλινικής, διαμορφώνουν τη διαγνωστική συλλογιστική και ως εκ τούτου την έγκαιρη αναγνώριση ή όχι του συνδρόμου.
Εκτός των άλλων, στη βιβλιογραφία δεν αναφέρονται ξεκάθαρες διαφορές στην επίπτωση ανάμεσα στα δύο φύλα ή την εθνικότητα. Όσον αφορά την επίπτωση στα δύο φύλα για τον παιδιατρικό πληθυσμό αυτή υπολογίζεται 1:1, ενώ για τους ενήλικες φαίνεται να υπάρχει ελαφρά υπεροχή στα άρρενα άτομα με αναλογία ~2:1. Όσον αφορά την επιβίωση ασθενών με HLH, είναι απαραίτητο να τονίσουμε την υψηλή θνητότητα του συνδρόμου, η οποία μπορεί να εξαρτάται από το υποκείμενο αίτιο, αλλά αγγίζει περίπου το 40% (!) (2- 19.5% όταν το υποκείμενο αίτιο είναι κάποια αυτοάνοση κατάσταση, έως 72% σε μερικά λεμφώματα).
Αιτιολογία
Το αιμοφαγοκυτταρικό σύνδρομο, μπορεί να είναι συγγενές (πρωτοπαθές)-κυρίως σε παιδιά ή επίκτητο (δευτεροπαθές).
-Συγγενές:
- Οικογενές HLH (Μεταλλάξεις γονιδίων Perforin- PRF1, Munc13-4 -UNC13D, Syntaxin 11-STX11, and Munc18-2 -STXBP2)
- Σύνδρομα ανοσοανεπάρκειας (Griscelli syndrome type 2 -GS2, X-linked lymphoproliferative -XLP και Chédiak-Higashi -CHS)
-Επίκτητο:
- Λοιμώξεις:
-Ιοί ( EBV, CMV, HAV, HBV, HCV, HSV, HHV-8, HIV, Parvo, Ιλαρά, Influenza, Sars-Cov2),
-Βακτήρια (Brucella sp., Bartonella henselae, Mycobacterium tb., κ.ά. Gram (-))
-Παράσιτα (Leishmania sp. (κυρίως L.donovani, L.infantum στη σπλαγχνική λεϊσμανίαση) , Plasmodium, Histoplasma, Toxoplasma, P. jiroveci)
-Μύκητες
- Αυτοάνοσα νοσήματα (Συστηματικός Ερυθηματώδης Λύκος, Ρευματοειδής Αρθρίτιδα (ιδιαίτερα στη Νεανική ΡΑ), Νόσος Still, Σαρκοείδωση, Οζώδης πολυαρτηρίτιδα, Σ. Sjogren’s)
Tip: Κλασικά, η HLH που εμφανίζεται στο πλαίσιο αυτοάνοσων διαταραχών, χαρακτηρίζεται ως σύνδρομο ενεργοποίησης μακροφάγων (Macrophage Activation Syndrome, MAS). Χαρακτηριστικά είναι εδώ, τα παραδείγματα της νεανικής ΡΑ και της νόσου Still στους ενήλικες.
- Νεοπλασματικά νοσήματα (Non Hodgkin Λεμφώματα, Λευχαιμίες)
- Ανοσοκαταστολή/ Μεταμόσχευση
- Σύνδρομο HELLP κατά την εγκυμοσύνη (Hemolysis /Elevated Liver enzymes/Low Plts count) (σχετικά σπάνια)
Όσον αφορά το σχετιζόμενο με τη λεϊσμανίαση αιμοφαγοκυτταρικό σύνδρομο, αξίζει να αναφερθεί ότι η χημειοθεραπευτική αγωγή (με λιποσωμιακή αμφοτερικίνη), έναντι του πρωτοπαθούς αιτίου μπορεί να συμβάλλει στον έλεγχο του συνδρόμου. Αν και για τις ενδημικές περιοχές του παρασίτου, αναφέρεται στη βιβλιογραφία ότι η παρόμοια κλινική εικόνα της σπλαγχνικής λεϊσμανίασης και του αιμοφαγοκυτταρικού συνδρόμου οδηγεί σε καθυστερημένη διάγνωση. Άλλωστε, είναι αναγκαίο σε αυτές τις περιοχές να γίνεται έλεγχος έναντι του παρασίτου, πριν από την έναρξη της ανοσοκατασταλτικής αγωγής.
Παθοφυσιολογία
Το αιμοφαγοκυτταρικό σύνδρομο χαρακτηρίζεται κυρίως από διαταραχή λειτουργίας του έμφυτου ανοσιακού συστήματος και πιο συγκεκριμένα, των κυττάρων φυσικών φονέων (ΝΚ), αλλά και των κυτταροτοξικών CD8+ T-λεμφοκυττάρων.

Σε έναν υγιή οργανισμό, τα κύτταρα αυτά παράγουν δύο κατηγορίες κυτταρολυτικών ενζύμων: την περφορίνη (perforin) και τα κοκκιοένζυμα (granzymes). Αυτά πακετάρονται μέσα σε κοκκία, τα οποία στη συνέχεια απελευθερώνονται στη σύναψη που δημιουργείται μεταξύ των κυττάρων του ανοσοποιητικού συστήματος και του κυττάρου στόχου. Η περφορίνη, επιπλέον, έχει την ιδιότητα να σχηματίζει ασταθείς πόρους στην επιφάνεια του κυττάρου στόχου, με αποτέλεσμα την εισροή ισχυρών πρωτεολυτικών ενζύμων εντός αυτού και τελικά την αποδόμησή του.

Σε ασθενείς με HLH , αυτή η διεργασία αναστέλλεται μέσω συγκεκριμένων γενετικών μεταλλάξεων ή ακόμη και επίκτητων, λόγω κάποιου ερεθίσματος που διεγείρει το ανοσιακό σύστημα (π.χ. κύτταρα μολυσμένα από ιό ή κακοήθη κύτταρα). Αυτή η αναποτελεσματική αλληλεπίδραση ανάμεσα στα NK κύτταρα, τα κυτταροτοξικά CD 8+ T-κύτταρα και τα κύτταρα στόχους, οδηγεί σε ένα φαύλο κύκλο ανεξέλεγκτης φλεγμονώδους απάντησης. Το αποτέλεσμα αυτής είναι να επιστρατεύονται ολοένα και περισσότερα κυτταροτοξικά Τ-κύτταρα με συνοδό υπερπαραγωγή κυτταροκινών, η οποία με τη σειρά της οδηγεί σε διάχυτη ενεργοποίηση μακροφάγων, αιμοφαγοκυττάρωση και εκδηλώσεις πολυσυστηματικής οργανικής ανεπάρκειας, στοιχεία τα οποία συνιστούν εν τέλει το αιμοφαγοκυτταρικό σύνδρομο. Με άλλα λόγια τα κυτταροτοξικά CD 8+ T-κύτταρα και τα NK κύτταρα δεν έχουν “φρένο”, και είναι μόνιμα ενεργοποιημένα.

https://images.app.goo.gl/KQDsghUowKvexezZA
Κλινική εικόνα
Η κλινική εκδήλωση του συνδρόμου, ενδέχεται να περιλαμβάνει:
–Εμπύρετο
–Σπληνομεγαλία/Ηπατομεγαλία
–Λεμφαδενική διόγκωση
-Ίκτερο
-Ωχρότητα
-Εξάνθημα
-Πετέχειες
-Εκχυμώσεις
-Οιδήματα
–Νευρολογικά συμπτώματα λόγω προσβολής του ΚΝΣ, όπως ευερεθιστότητα, αλλαγή του επιπέδου συνείδησης, προσβολή εγκεφαλικών συζυγιών, σπασμούς, επιληψία, περιφερική νευροπάθεια ή ακόμη και κώμα
–Αναπνευστική ανεπάρκεια
–Αιμοδυναμική αστάθεια
Εργαστηριακά ευρήματα
–Πανκυτταροπενία, πιο συχνά: αναιμία και θρομβοπενία, λόγω κατανάλωσης στην αιμοφαγοκυττάρωση και της καταστροφής από τις κυτταροκίνες TNF-a και IFN-γ.
–Υπερφερριτιναιμία, κυρίως λόγω παθητικής έκλυσης από την κυτταρική καταστροφή και τη φαγοκυττάρωση, αλλά και λόγω γονιδιακής ενεργοποίησης από τον TNF-a. Μέσα σε λίγες ώρες από την εκδήλωση του συνδρόμου, μπορεί να αυξηθεί >10.000 mg/dl (se 90%, sp 96%).
–Υπερτριγλυκεριδαιμία, λόγω καταστολής της λιποπρωτεϊνικής λιπάσης από τον TNF-a.
–Υποινωδογοναιμία, λόγω διέγερσης του πλασμινογόνου από ενεργοποιημένα μακροφάγα και πτώση ταχύτητας καθίζησης.
–Τρανσαμινασαιμία ή /και Υπερχολερυθριναιμία.
–Υπολευκωματιναιμία.
–↑ LDH ορού >1000 IU/L.
–↑ Διαλυτός υποδοχέας της IL-2 (sCD25).
–ΕΥΡΗΜΑΤΑ ΕΝΥ:
- Μέτρια πλειοκυττάρωση ~20−80 λεμφοκύτταρα/μL (κυρίως Τ-λεμφοκύτταρα).
- Αύξηση λευκώματος σε 0.5−1 g/L και
- Αιμοφαγοκυττάρωση στα ιστιοκύτταρα-μακροφάγα του ΕΝΥ.
Tip: Άρα αν ο ασθενής έχει πυρετό, ηπατοσπληνομεγαλία, λεμφαδενοπάθεια, σύγχυση, σοκ και ευρήματα όπως αναιμία, θρομβοπενία, ουδετεροπενία, αυξημένα ηπατικά και φερριτίνη = σκέψου αιμοφαγοκυτταρικό!
Διάγνωση
Η HLH διαγιγνώσκεται μέσα από μια σειρά σημείων, συμπτωμάτων, αλλά και εργαστηριακών ευρημάτων. Συχνά, η παρουσία μη ειδικών συμπτωμάτων και ο υψηλός δείκτης κλινικής υποψίας είναι απαραίτητα για την άμεση επιβεβαίωση και θεραπεία του συνδρόμου. Η πρώιμη διάγνωση είναι αναγκαία, καθώς η καθυστερημένη έναρξη θεραπείας μπορεί να οδηγήσει ακόμη και σε θάνατο. Το 2004, η “The Histiocyte Society” πρότεινε τη θέσπιση ενός συνδυασμού κριτηρίων, μέσω του οποίου θα διαγιγνώσκονται ασθενείς με HLH (βλ. πίνακα). Αρκεί να πληρούνται 5 από τα 8 κριτήρια για την επιβεβαίωση της διάγνωσης. Κατά την αρχική εκτίμηση, οι ασθενείς μπορεί να μην πληρούν όλα αυτά τα κριτήρια, καθιστώντας τη διάγνωση μια πραγματική πρόκληση.
Επίσης, είναι πολύ σημαντικό να τονίσουμε ότι αυτά τα κριτήρια, αναπτύχθηκαν αρχικά για τη διάγνωση της HLH σε παιδιατρικό πληθυσμό, αλλά χρησιμοποιούνται πλέον ευρέως και σε ενήλικες ασθενείς. Μέχρι στιγμής πάντως, δεν υπάρχουν διεθνώς επικαιροποιημένα κριτήρια μόνο για ενήλικες.
Διαγνωστικά κριτήρια
| Μοριακή διάγνωση συμβατή με HLH |
| Τουλάχιστον 5 από τα ακόλουθα 8 κλινικοεργαστηριακά κριτήρια |
| 1. Πυρετός |
| 2. Σπληνομεγαλία |
| 3. Κυτταροπενία (> 2 σειρές) •Hb < 9 gr/dl •PLTs < 100.000/μl •PMN < 1.000/μl |
| 4. Υπερτριγλυκεριδαιμία (>265 mg/dl) ή υποϊνωδογοναιμία (<150 mg/dl) |
| 5. Αιμοφαγοκυττάρωση στο μυελό ή σε βιοψία ήπατος ή λεμφαδένα* |
| 6. Φεριττίνη ορού > 500 μg/l |
| 7. ↑ Διαλυτός υποδοχέας της IL-2 (sCD25) (> 2400 U/ml)** |
| 8. ↓ δραστηριότητα των κυττάρων ΝΚ |
*Η παρουσία αιμοφαγοκυττάρωσης στο μυελό των οστών είναι χρήσιμη για τη διάγνωση της HLH, αλλά η απουσία της ΔΕΝ την αποκλείει.
** Η αύξηση του sIL‐2 υποδοχέα >2515 U/ml, έχει ευαισθησία se 100% και ειδικότητα sp 72.5% για τη διάγνωση.
Tip: Βασική διαφοροδιάγνωση πρέπει να τεθεί ανάμεσα σε αιμοφαγοκυτταρικό και σήψη, TTP/HUS, DRESS. Το αιμοφαγοκυτταρικό ξεχωρίζει από την σήψη ως προς την Υπερτριγλυκεριδαιμία, Υπερφερριτιναιμία, Υποινωδογοναιμία και μείωση ΤΚΕ.
Θεραπεία
Ο ακρογωνιαίος λίθος της θεραπείας του αιμοφαγοκυτταρικού συνδρόμου, είναι η ανοσοκαταστολή και η κυτταροτοξική θεραπεία, οι οποίες παρ’όλα αυτά αντενδείκνυνται σε ασθενείς με σοβαρή λοίμωξη. Όπως αναφέρθηκε και παραπάνω, είναι σημαντικό πριν την έναρξη θεραπείας να υπάρχει ισχυρή κλινική υπόνοια υπέρ του συνδρόμου, ακόμη και αν δεν πληρούνται τα απαραίτητα διαγνωστικά κριτήρια. Δηλαδή, θα μπορούσαμε να ξεκινήσουμε θεραπεία με < 5 διαγνωστικά κριτήρια!
Το πρωτόκολλο HLH-94, εφαρμόστηκε για πρώτη φορά έπειτα από μια συλλογή δεδομένων που αφορούσε σε 113 παιδιατρικά κι επομένως, πρωτοπαθή περιστατικά HLH και περιελάμβανε τον εξής συνδυασμό ανοσοκατασταλτικών: δεξαμεθαζόνη, ετοποσίδη, κυκλοσπορίνη Α (CSA), καθώς και ενδοθηκικές εγχύσεις μεθοτρεξάτης σε επιλεγμένους ασθενείς που επρόκειτο να υποβληθούν σε μεταμόσχευση μυελού των οστών, με στόχο την καταστροφή των ενεργοποιημένων Τ-κυττάρων και την καταστολή της παραγωγής φλεγμονωδών κυτταροκινών. Τα αποτελέσματα ανέδειξαν ~ 55% ποσοστό επιβίωσης 3 έτη αργότερα και ~22% αντίστοιχο ποσοστό, για τα επόμενα 5 έτη. Ο τρόπος χορήγησης, φαίνεται στο διάγραμμα παρακάτω και περιλαμβάνει την αρχική θεραπεία διάρκειας 8 εβδομάδων με καθημερινή χορήγηση δεξαμεθαζόνης σε μειούμενη δόση ανά 2 εβδομάδες κι ακολούθως, ώσεις δεξαμεθαζόνης κάθε δεύτερη εβδομάδα για 3 ημέρες.
Ωστόσο, ενήλικες ασθενείς και ιδιαίτερα ηλικιωμένοι με χρόνιες συνοσηρότητες, είναι περισσότερο ευάλωτοι σε βλάβες τελικών οργάνων που σχετίζονται με την απελευθέρωση κυτταροκινών της HLH, αλλά και της χημειοθεραπείας που χρησιμοποιείται στο πρωτόκολλο HLH-94. Έτσι, η χορήγηση της ετοποσίδης, ως χημειοθεραπευτικός παράγοντας με υψηλή δραστικότητα έναντι της διαφοροποίησης των Τ-κυττάρων, θα πρέπει να γίνεται με συχνότητα μικρότερη από αυτή που προτείνει το HLH-94, κυμαινόμενη από 2 έως 1 φορές/εβδομάδα, με ή χωρίς μείωση τη δοσολογίας από 150 mg/m^2 σε 50-100 mg/m^2.
Όσον αφορά τη χορήγηση της κυκλοσπορίνης Α (CSA), η οποία έχει συσχετισθεί με ανεπιθύμητες ενέργειες* και αντενδείξεις, ιδιαίτερα κατά την έναρξη της θεραπείας, δεν θα πρέπει να χορηγείται πριν από την 3η εβδομάδα θεραπείας (συνήθως την 9η εβδομάδα).
*Οι πιο σημαντικές είναι: υπέρταση, νευροτοξικότητα (σύνδρομο οπίσθιας αναστρέψιμης εγκεφαλοπάθειας, Posterior reversible encephalopathy syndrome (PRES)), υπερκαλιαιμία, ηπατοτοξικότητα, δυσανεξία στη γλυκόζη και νεφροτοξικότητα. Χρειάζεται, επιπλέον, έλεγχος των επιπέδων κυκλοσπορίνης (<200 μg/L), καθώς πολλές από τις ανεπιθύμητες ενέργειες είναι δοσοεξαρτώμενες (!).
Τέλος, η ενδοθηκική θεραπεία με μεθοτρεξάτη προτείνεται μόνο για την περίπτωση προοδευτικά επιδεινούμενων νευρολογικών συμπτωμάτων, έπειτα από 2 εβδομάδες θεραπείας ή μη βελτίωσης παθολογικού ΕΝΥ.

La Rosée P, Horne A, Hines M, von Bahr Greenwood T, Machowicz R, Berliner N, Birndt S, Gil-Herrera J, Girschikofsky M, Jordan MB, Kumar A, van Laar JAM, Lachmann G, Nichols KE, Ramanan AV, Wang Y, Wang Z, Janka G, Henter JI. Recommendations for the management of hemophagocytic lymphohistiocytosis in adults. Blood. 2019 Jun 6;133(23):2465-2477. doi: 10.1182/blood.2018894618. Epub 2019 Apr 16. PMID: 30992265.
Στη συνέχεια, έγινε εφαρμογή του πρωτοκόλλου HLH-2004 το οποίο περιελάμβανε ασθενείς τόσο με πρωτοπαθή, όσο και με δευτεροπαθή HLH και συγκριτικά με το HLH-94, είχε την εξής διαφορά: προστέθηκε η κυκλοσπορίνη στο αρχικό θεραπευτικό σχήμα, ταυτόχρονα με ενδοθηκικές εγχύσεις στεροειδών σε ασθενείς με προυπάρχουσα παθολογία του νευρικού συστήματος. Ωστόσο, τα αποτελέσματα των μελετών δεν ανέδειξαν βελτίωση της συνολικής έκβασης των ασθενών κι ως εκ τούτου, το πρωτόκολλο HLH-94, συμπεριλαμβανομένης και της ετοποσίδης είναι αυτό που προτιμάται σε ενήλικες ασθενείς.
Παρακάτω, αναφέρεται μια περιεκτική ταξινόμηση η οποία περιλαμβάνει έναν ενδεικτικό αλγόριθμο, βάσει του οποίου στη σύγχρονη θεραπευτική προσέγγιση έχει θέση και η χορήγηση γ-σφαιρίνης (IVIG), αφενός λόγω της αντιφλεγμονώδους δράσης της και αφετέρου λόγω της αποκατάστασης της ομοιοστασίας του ανοσοποιητικού συστήματος.
Έτσι, αν συνοψίζαμε μια βασική διαχείριση ασθενούς με HLH, αυτή θα περιελάμβανε:
- Αντιμετώπιση του αιτιολογικού παράγοντα
- Κορτικοστεροειδή
- Γ-σφαιρίνη
Τέλος, στη σύγχρονη αντιμετώπιση ασθενών με HLH περιλαμβάνονται και νεότεροι βιολογικοί παράγοντες, όπως οι ανταγωνιστές των υποδοχέων IL-1 (IL-1Ra,KINERET® (anakinra)) και IL-6 (IL-6Ra, tocilizumab), κυρίως για περιπτώσεις αιμοφαγοκυτταρικού συνδρόμου επαγόμενου από αυτοάνοσα νοσήματα ή φάρμακα (π.χ. Checkpoint inhibitors).
Συνοπτικά:
Πρωτοπαθές HLH = πρωτόκολλο HLH-94 και μεταμόσχευση μυελού των οστών.
Δευτεροπαθές HLH = στεροειδή, HLH-94, Anakinra, IViG, Rituximab, Tacrolimus.

Προτεινόμενες πηγές
- Trottestam H, Beutel K, Meeths M, Carlsen N, Heilmann C, Pasić S, Webb D, Hasle H, Henter JI. Treatment of the X-linked lymphoproliferative, Griscelli and Chédiak-Higashi syndromes by HLH directed therapy. Pediatr Blood Cancer. 2009 Feb;52(2):268-72. doi: 10.1002/pbc.21790. PMID: 18937330.
- Eric Gars, Natasha Purington, Gregory Scott, Karen Chisholm, Dita Gratzinger, Beth A. Martin, Robert S. Ohgami. Bone marrow histomorphological criteria can accurately diagnose hemophagocytic lymphohistiocytosis. Haematologica 2018;103(10):1635-1641; https://doi.org/10.3324/haematol.2017.186627.
- Ishii, E. (2016) ‘Hemophagocytic lymphohistiocytosis in children: Pathogenesis and treatment’, Frontiers in Pediatrics, 4. doi:10.3389/fped.2016.00047.
- Jordan MB. Hemophagocytic lymphohistiocytosis: A disorder of T cell activation, immune regulation, and distinctive immunopathology. Immunol Rev. 2023 Dec 15. doi: 10.1111/imr.13298. Epub ahead of print. PMID: 38100247.
- Neycheva S, Oparanov B, Kamburova A, Karalilova R, Stoeva V. Hemophagocytic Lymphohistiocytosis Triggered by Leishmaniasis: A Case Report and Literature Review. Am J Case Rep. 2021 Oct 6;22:e933012. doi: 10.12659/AJCR.933012. PMID: 34613957; PMCID: PMC8503795.
- Konkol S, Rai M. Lymphohistiocytosis. [Updated 2023 Mar 27]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK557776/
- Hempel A, Manzoor F, Petrescu D. Hemophagocytic lymphohistiocytosis secondary to unrecognized Bartonella henselae infection: a case report. Trop Dis Travel Med Vaccines. 2023 Sep 25;9(1):14. doi: 10.1186/s40794-023-00200-1. PMID: 37743475; PMCID: PMC10518968.
- Sarkissian S, Khan Y, Farrell D, Constable D, Brem E. Hemophagocytic lymphohistiocytosis in the setting of HELLP Syndrome. Clin Case Rep. 2018; 6: 2466–2470. https://doi.org/10.1002/ccr3.1828
- Dilibe A, Ugoala O S, Evbayekha E O, et al. (March 01, 2023) Macrophage Activation Syndrome (MAS): A Case Report and Narrative Review. Cureus 15(3): e35670. doi:10.7759/cureus.35670
- Δανά Ελένη, Αιμοφαγοκυτταρικό σύνδρομο, Ογκολογική Κλινική Παιδιών & Εφήβων, Παίδων, Μητέρα
- Alison M. Schram, Nancy Berliner; How I treat hemophagocytic lymphohistiocytosis in the adult patient. Blood 2015; 125 (19): 2908–2914. doi: https://doi.org/10.1182/blood-2015-01-551622
- La Rosée P, Horne A, Hines M, von Bahr Greenwood T, Machowicz R, Berliner N, Birndt S, Gil-Herrera J, Girschikofsky M, Jordan MB, Kumar A, van Laar JAM, Lachmann G, Nichols KE, Ramanan AV, Wang Y, Wang Z, Janka G, Henter JI. Recommendations for the management of hemophagocytic lymphohistiocytosis in adults. Blood. 2019 Jun 6;133(23):2465-2477. doi: 10.1182/blood.2018894618. Epub 2019 Apr 16. PMID: 30992265.
- La Marle, S.; Richard-Colmant, G.; Fauvernier, M.; Ghesquières, H.; Hot, A.; Sève, P.; Jamilloux, Y. Mortality and Associated Causes in Hemophagocytic Lymphohistiocytosis: A Multiple-Cause-of-Death Analysis in France. J. Clin. Med. 2023, 12, 1696. https://doi.org/10.3390/jcm12041696
- HLH I Hemophagocytic Lymphohistiocytosis – cause, pathophysiology, investigation and treatment
Συντελεστές/Δημιουργοί: Τα Μέλη της Ομάδας του Klinikal GOMED
- Συγγραφέας Περιεχομένου: Παπαθανασίου Σοφία, MD, Ειδικευόμενη Παθολογίας
- Reviewer: Κυριακούλη Ιωάννα, MD, Ειδικευόμενη Καρδιολογίας
- Coordinator: Σικόλας Αριστείδης, MD, Ειδικευόμενος Καρδιολογίας